Indicator species analysis
Miquel De Cáceres, CREAF
2025-02-20
Source:vignettes/IndicatorSpeciesAnalysis.Rmd
IndicatorSpeciesAnalysis.RmdIntroduction
Determining the occurrence or abundance of a small set of indicator species, as an alternative to sampling the entire community, has been particularly useful in longterm environmental monitoring for conservation or ecological management. Species are chosen as indicators if they:
- reflect the biotic or abiotic state of the environment;
- provide evidence for the impacts of environmental change; or
- predict the diversity of other species, taxa or communities within an area.
In this tutorial we will show how to use the functions included in
package indicspecies to conduct indicator species analysis.
This package was originally created as a supplementary material to De Cáceres and Legendre (2009), but has been
developing since then and now indicspecies updates are
distributed from CRAN and GitHub repositories.
Before doing anything else, we need to load the functions of the package:
library(indicspecies)
#> Loading required package: permuteData required for indicator species analysis
Indicator species are often determined using an analysis of the relationship between the species occurrence or abundance values from a set of sampled sites and the classification of the same sites into site groups, which may represent habitat types, community types, disturbance states, etc. Thus, there are two data elements in an indicator species analysis: (1) the community data matrix; and (2) the vector that describes the classification of sites into groups.
The community data matrix
This is a matrix (or a data frame) with sites in
rows and species in columns. Normally, we will
use functions like read.table() or read.csv()
to read our data set from a file. In this example we load our example
dataset into the workspace using:
data(wetland)The wetland data set describes the vegetation of the Adelaide river alluvial plain (Australia), as sampled by Bowman and Wilson (1987). It contains the abundance values of 33 species (columns) in 41 sites (rows).
Defining the classification of sites
In order to run an indicator species analysis we need a vector
containing the classification of the sites into groups. The
intepretation of these site groups is left to the user. Like community
data, site classifications can be read from a data file. A vector of
site groups can be also created manually, for example, using the R
functions c() and rep():
groups <- c(rep(1, 17), rep(2, 14), rep(3,10))
groups
#> [1] 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 2 2 2 2 2 2 2 2 2 2 2 2 2 2 3 3 3 3 3 3 3
#> [39] 3 3 3Alternatively, one can obtain a classification using non-hierarchical cluster analysis:
wetkm <- kmeans(wetland, centers=3)
groupskm <- wetkm$cluster
groupskm
#> 5 8 13 4 17 3 9 21 16 14 2 15 1 7 10 40 23 25 22 20 6 18 12 39 19 11
#> 2 2 2 2 2 2 2 2 2 2 2 2 2 2 2 1 1 1 1 1 1 1 1 1 1 1
#> 30 34 28 31 26 29 33 24 36 37 41 27 32 35 38
#> 3 3 3 3 1 1 1 3 3 3 3 3 3 2 2If the site classification vector is obtained independently of species data, the significance of statistical tests carried out on the indicator species will be meaningful. For example, one could classify the sites using environmental data before indicator species analysis. An example is found in Borcard, Gillet, and Legendre (2011).
Indicator species analysis using multipatt()
Function multipatt() is the most commonly used function
of indicspecies. It allows determining lists of species
that are associated to particular groups of sites (or combinations of
those). Once we have the two data components mentioned in the previous
section, we are ready to run an indicator species analysis using
multipatt().
Indicator Value analysis with site group combinations
When the aim is to determine which species can be used as indicators
of certain site group an approach commonly used in ecology is the
Indicator Value (Dufrêne and Legendre
1997). These authors defined an Indicator Value (IndVal) index to
measure the association between a species and a site group. The method
of Dufrêne and Legendre (1997) calculates
the IndVal index between the species and each site group and then looks
for the group corresponding to the highest association value. Finally,
the statistical significance of this relationship is tested using a
permutation test. IndVal is the default index used to measure the
association between a species and a group of sites in
multipatt(). However, by default multipatt()
uses an extension of the original Indicator Value method, because the
function looks for indicator species of both individual site groups
and combinations of site groups, as explained in De Cáceres, Legendre, and Moretti (2010).
Indicator species analysis (with site group combinations) can be run using:
As mentioned before, by default multipatt() uses the
IndVal index (func = "IndVal.g") as test statistic.
Actually, the square root of IndVal is returned by the
multipatt() function. The option
control = how(nperm=999) allows choosing the number of
random permutations required for the permutational test (this number
affects the precision of the p-value). Function how() from
the permute package allows defining more complex
permutational designs.
Displaying the results
When the indicator species analysis is completed, we can obtain the list of indicator species for each site group (or site group combination) using:
summary(indval)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 10
#> Number of species associated to 1 group: 6
#> Number of species associated to 2 groups: 4
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 3
#> stat p.value
#> Ludads 0.907 0.001 ***
#> Orysp. 0.823 0.002 **
#> Psespi 0.602 0.022 *
#>
#> Group 3 #sps. 3
#> stat p.value
#> Pancam 0.910 0.001 ***
#> Eupvac 0.724 0.003 **
#> Cynarc 0.602 0.009 **
#>
#> Group 1+2 #sps. 1
#> stat p.value
#> Elesp. 0.741 0.007 **
#>
#> Group 2+3 #sps. 3
#> stat p.value
#> Melcor 0.876 0.001 ***
#> Phynod 0.715 0.008 **
#> Echell 0.651 0.011 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1In our wetland community data, ‘Ludads’ is strongly and significantly associated with Group 1, whereas ‘Pancam’ would be a good indicator of Group 3. In addition, there are some species whose patterns of abundance are more associated with a combination of groups. For example, ‘Melcor’ is strongly associated with the combination of Groups 2 and 3.
It is important to stress that the indicator species analysis is conducted for each species independently, although the results are often summarized for all species. User should bear in mind possible problems of multiple testing when making community-level statements such as ‘the number of indicator species is X’ (De Cáceres and Legendre 2009; Legendre and Legendre 2012).
Examining the indicator value components
If the association index used in multipatt() is
func = "IndVal" or func = "IndVal.g", one can
also inspect the indicator value components when displaying the results.
Indeed, the indicator value index is the product of two components,
called ‘A’ and ‘B’ (Dufrêne and Legendre 1997; De
Cáceres and Legendre 2009):
- Component ‘A’ is sample estimate of the probability that the surveyed site belongs to the target site group given the fact that the species has been found. This conditional probability is called the specificity or positive predictive value of the species as indicator of the site group.
- Component ‘B’ is sample estimate of the probability of finding the species in sites belonging to the site group. This second conditional probability is called the fidelity or sensitivity of the species as indicator of the target site group.
To display the indicator value components ‘A’ and ‘B’ one simply uses:
summary(indval, indvalcomp=TRUE)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 10
#> Number of species associated to 1 group: 6
#> Number of species associated to 2 groups: 4
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 3
#> A B stat p.value
#> Ludads 1.0000 0.8235 0.907 0.001 ***
#> Orysp. 0.6772 1.0000 0.823 0.002 **
#> Psespi 0.8811 0.4118 0.602 0.022 *
#>
#> Group 3 #sps. 3
#> A B stat p.value
#> Pancam 0.8278 1.0000 0.910 0.001 ***
#> Eupvac 0.6546 0.8000 0.724 0.003 **
#> Cynarc 0.7241 0.5000 0.602 0.009 **
#>
#> Group 1+2 #sps. 1
#> A B stat p.value
#> Elesp. 1.0000 0.5484 0.741 0.007 **
#>
#> Group 2+3 #sps. 3
#> A B stat p.value
#> Melcor 0.8764 0.8750 0.876 0.001 ***
#> Phynod 0.8752 0.5833 0.715 0.008 **
#> Echell 0.9246 0.4583 0.651 0.011 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1This gives us additional information about why species can be used as indicators. For example, ‘Ludads’ is a good indicator of Group 1 because it occurs in sites belonging to this group only (i.e., A = 1.0000), although not all sites belonging to Group 1 include the species (i.e., B = 0.8235). In contrast, ‘Pancam’ can be used to indicate Group 3 because it appears in all sites belonging to this group (i.e., B = 1.0000) and it is largely (but not completely) restricted to it (i.e., A = 0.8278).
Inspecting the indicator species analysis results for all species
In our previous calls to summary() only the species that
were significantly associated with site groups (or site group
combinations) were shown. One can display the result of the indicator
species analysis for all species, regardless of whether the
permutational test was significant or not. This is done by changing the
significance level in the summary:
summary(indval, alpha=1)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 1
#>
#> Total number of species: 33
#> Selected number of species: 29
#> Number of species associated to 1 group: 21
#> Number of species associated to 2 groups: 8
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 5
#> stat p.value
#> Ludads 0.907 0.001 ***
#> Orysp. 0.823 0.002 **
#> Psespi 0.602 0.022 *
#> Polatt 0.420 0.154
#> Casobt 0.243 1.000
#>
#> Group 2 #sps. 6
#> stat p.value
#> Aesind 0.445 0.233
#> Alyvag 0.335 0.446
#> Abefic 0.267 0.577
#> Poa2 0.267 0.583
#> Poa1 0.267 0.609
#> Helcri 0.267 0.609
#>
#> Group 3 #sps. 10
#> stat p.value
#> Pancam 0.910 0.001 ***
#> Eupvac 0.724 0.003 **
#> Cynarc 0.602 0.009 **
#> Abemos 0.447 0.058 .
#> Merhed 0.402 0.211
#> Ludoct 0.316 0.247
#> Passcr 0.316 0.233
#> Dendio 0.316 0.237
#> Physp. 0.316 0.249
#> Goopur 0.316 0.249
#>
#> Group 1+2 #sps. 2
#> stat p.value
#> Elesp. 0.741 0.007 **
#> Carhal 0.402 0.427
#>
#> Group 2+3 #sps. 6
#> stat p.value
#> Melcor 0.876 0.001 ***
#> Phynod 0.715 0.008 **
#> Echell 0.651 0.011 *
#> Echpas 0.584 0.273
#> Cyprot 0.500 0.067 .
#> Ipocop 0.354 0.325
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Parameter alpha is by default set to
alpha = 0.05, and hides all species association that are
not significant at this level. By setting alpha = 1 we say
we want to display the group to which each species is associated,
regardless of whether the association significant or not. However, note
that in our example we obtain the results of 29 (21+8) species. As there
are 33 species in the data set, there are still four species missing in
this summary. This happens because those species have their highest
IndVal value for the set of all sites. In other words, those species
occur in sites belonging to all groups. The association with the set of
all sites cannot be statistically tested, because there is no external
group for comparison. In order to know which species are those, one has
to inspect the object sign returned by
multipatt():
indval$sign
#> s.1 s.2 s.3 index stat p.value
#> Abefic 0 1 0 2 0.2672612 0.577
#> Merhed 0 0 1 3 0.4019185 0.211
#> Alyvag 0 1 0 2 0.3347953 0.446
#> Pancam 0 0 1 3 0.9098495 0.001
#> Abemos 0 0 1 3 0.4472136 0.058
#> Melcor 0 1 1 6 0.8757059 0.001
#> Ludoct 0 0 1 3 0.3162278 0.247
#> Eupvac 0 0 1 3 0.7236825 0.003
#> Echpas 0 1 1 6 0.5842649 0.273
#> Passcr 0 0 1 3 0.3162278 0.233
#> Poa2 0 1 0 2 0.2672612 0.583
#> Carhal 1 1 0 4 0.4016097 0.427
#> Dendio 0 0 1 3 0.3162278 0.237
#> Casobt 1 0 0 1 0.2425356 1.000
#> Aesind 0 1 0 2 0.4447093 0.233
#> Cyprot 0 1 1 6 0.5000000 0.067
#> Ipocop 0 1 1 6 0.3535534 0.325
#> Cynarc 0 0 1 3 0.6017217 0.009
#> Walind 1 1 1 7 0.4938648 NA
#> Sessp. 1 1 1 7 0.6984303 NA
#> Phynod 0 1 1 6 0.7145356 0.008
#> Echell 0 1 1 6 0.6509834 0.011
#> Helind 1 1 1 7 0.6984303 NA
#> Ipoaqu 1 1 1 7 0.4938648 NA
#> Orysp. 1 0 0 1 0.8229074 0.002
#> Elesp. 1 1 0 4 0.7405316 0.007
#> Psespi 1 0 0 1 0.6023402 0.022
#> Ludads 1 0 0 1 0.9074852 0.001
#> Polatt 1 0 0 1 0.4200840 0.154
#> Poa1 0 1 0 2 0.2672612 0.609
#> Helcri 0 1 0 2 0.2672612 0.609
#> Physp. 0 0 1 3 0.3162278 0.249
#> Goopur 0 0 1 3 0.3162278 0.249After accessing the object indval$sign, we know that the
four species whose highest IndVal corresponded to the set of all sites
were ‘Valind’, ‘Sessp.’, ‘Helind’ and ‘Ipoaqu’, as indicated by the
missing values in the p.value column of the data frame. The
first columns of indicate (with ones and zeroes) which site groups were
included in the combination preferred by the species. Then, the column
indicates the index of the site group combination (see subsection
Excluding site group combinations in multipatt() below).
The remaining two columns are the association statistic and the p-value
of the permutational test.
Analyzing species ecological preferences with correlation indices
Several other indices can be used to analyze the association between
a species and a group of sites (De Cáceres and
Legendre 2009). Diagnostic (or indicator) species are an
important tool in vegetation science, because these species can be used
to characterize and indicate specific plant community types. A statistic
commonly used to determine the association (also known as
fidelity, not to be confounded with the indicator value
component) between species and vegetation types is Pearson’s phi
coefficient of association (Chytrý et al.
2002). This coefficient is a measure of the correlation between
two binary vectors. It is possible to calculate the phi coefficient in
multipatt() after transforming our community data to
presence-absence:
wetlandpa <- ifelse(wetland>0,1,0)
phi <- multipatt(wetlandpa, groups, func = "r",
control = how(nperm=999)) What would be the association index if we had used abundance values
instead of presence and absences (i.e. wetland instead of
wetlandpa)? The abundance-based counterpart of the phi
coefficient is called the point biserial correlation
coefficient.
It is a good practice to correct the phi coefficient for the fact
that some groups have more sites than others (Tichý and Chytrý 2006). To do that, we need to
use func = "r.g" instead of func = "r":
Remember that the default association index of
multipatt() is func = "IndVal.g", which also
includes “.g”. In fact, the Indicator Value index defined by Dufrêne and Legendre (1997) already incorporated
a correction for unequal group sizes. It is possible to avoid this
correction by calling multipatt() with
func = "IndVal". However, in general we recommend using
either func = "IndVal.g" or func = "r.g" for
indicator species analysis.
Indicator value and correlation indices usually produce similar results. Indeed, if we display the results of the phi coefficient of association we see that they are qualitatively similar to those of IndVal:
summary(phi)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: r.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 10
#> Number of species associated to 1 group: 8
#> Number of species associated to 2 groups: 2
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 3
#> stat p.value
#> Ludads 0.870 0.001 ***
#> Orysp. 0.668 0.001 ***
#> Psespi 0.413 0.012 *
#>
#> Group 2 #sps. 1
#> stat p.value
#> Phynod 0.436 0.021 *
#>
#> Group 3 #sps. 4
#> stat p.value
#> Pancam 0.748 0.001 ***
#> Eupvac 0.537 0.003 **
#> Cynarc 0.492 0.012 *
#> Abemos 0.378 0.041 *
#>
#> Group 1+2 #sps. 1
#> stat p.value
#> Elesp. 0.538 0.004 **
#>
#> Group 2+3 #sps. 1
#> stat p.value
#> Melcor 0.612 0.001 ***
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Nevertheless, there are some differences between indicator values and correlation indices (De Cáceres, Font, and Oliva 2008; De Cáceres and Legendre 2009). Correlation indices are used for determining the ecological preferences of species among a set of alternative site groups or site group combinations. Indicator value indices are used for assessing the predictive values of species as indicators of the conditions prevailing in site groups, e.g. for field determination of community types or ecological monitoring.
An advantage of the phi and point biserial coefficients is that they
can take negative values. When this happens, the value of the index is
expressing the fact that a species tends to ‘avoid’ particular
environmental conditions. We will find negative association values if we
inspect the strength of association in the results of
multipatt() when these coefficients are used:
round(head(phi$str),3)
#> 1 2 3 1+2 1+3 2+3
#> Abefic -0.110 0.221 -0.110 0.110 -0.221 0.110
#> Merhed -0.223 -0.047 0.270 -0.270 0.047 0.223
#> Alyvag -0.024 0.214 -0.190 0.190 -0.214 0.024
#> Pancam -0.585 -0.163 0.748 -0.748 0.163 0.585
#> Abemos -0.189 -0.189 0.378 -0.378 0.189 0.189
#> Melcor -0.612 0.142 0.470 -0.470 -0.142 0.612In contrast, indicator values are always non-negative:
round(head(indval$str),3)
#> 1 2 3 1+2 1+3 2+3 1+2+3
#> Abefic 0.000 0.267 0.000 0.180 0.000 0.204 0.156
#> Merhed 0.000 0.117 0.402 0.079 0.245 0.354 0.271
#> Alyvag 0.113 0.335 0.000 0.311 0.089 0.256 0.271
#> Pancam 0.038 0.230 0.910 0.183 0.589 0.781 0.625
#> Abemos 0.000 0.000 0.447 0.000 0.272 0.289 0.221
#> Melcor 0.191 0.509 0.739 0.484 0.610 0.876 0.796The fact that correlation indices take negative values means that
they can be used to detect negative associations (i.e. site
groups or site group combinations that are statistically avoided by the
species). Function multipatt() can be used for this purpose
without modification, if all groups combinations are explored. The
reason is that strength of correlation for a given site group
combination has always the same absolute value than the correlation for
the complementary combination (you can verify this in the table above
displaying the strength of association results for phi).
This entails that the site group combination that maximizes positive
association is the complement of the site group combination that
minimizes (i.e. maximizes the negative value) negative association. For
example, in species ‘Pancam’ the maximum positive phi correlation is for
Group 3 and the minimum negative phi correlation is for the combination
of Group 1 and Group 2 (i.e. the complement of Group 3). Hence, one
could use the complement of site group combinations indicated by
multipatt() for positive associations as results for
negative associations. Following the same example, species ‘Pancam’
would be negatively associated to the combination of Group 1 and Group 2
with the same p-value as obtained for its positive association with
Group 3.
Alternatively, one can allow multipatt() to explicitly
look for both negative and positive associations by
setting allow.negative = TRUE:
In this case, multipatt() will keep the association with
highest strength in absolute value. Since ties can occur (in absolute
value) between a group and its complementary, as explained above, the
function will in this case keep the association corresponding to group
combination of the smallest order.
summary(phi)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: r.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 9
#> Number of species associated to 1 group: 9
#> Number of species associated to 2 groups: 0
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 4
#> stat p.value
#> Ludads 0.870 0.001 ***
#> Orysp. 0.668 0.001 ***
#> Melcor -0.612 0.001 ***
#> Psespi 0.413 0.020 *
#>
#> Group 2 #sps. 1
#> stat p.value
#> Phynod 0.436 0.015 *
#>
#> Group 3 #sps. 4
#> stat p.value
#> Pancam 0.748 0.001 ***
#> Elesp. -0.538 0.005 **
#> Eupvac 0.537 0.004 **
#> Cynarc 0.492 0.013 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Note that in this case, species ‘Melcor’ and ‘Elesp’ appear as negatively associated to single groups, whereas in the previous call (not allowing negative associations) they were reported as positively associated to the complementary group combination.
Unlike with indicator value coefficients, the set of all sites can never be considered with the phi or point biserial coefficients, because these coefficients always require a set of sites for comparison, besides the target site group or site group combination of interest.
Excluding site group combinations in multipatt()
There are two situations where site group combinations may be excluded from the analysis:
- When conducting indicator species analysis, it may happen that some combinations of site groups are difficult to interpret ecologically. In those cases, we may decide to exclude those combinations from the analysis, so our species may appear associated to other (more interpretable) ecological conditions.
- The combinations to be explored will need to be restricted if the initial number of groups is large (e.g. more than 15), because the number of possible combinations grows exponentially and calculations will become computationally unfeasible if combinations are not restricted.
There are three ways to restrict the site group combinations to be
considered in multipatt(), as detailed in the following
subsections.
Indicator species analysis without site groups combinations
The original Indicator Value method of Dufrêne
and Legendre (1997) did not consider combinations of site groups.
In other words, the only site group combinations permitted in the
original method were singletons. When using multipatt() it
is possible to avoid considering site group combinations, as in the
original method, by using duleg = TRUE:
indvalori <- multipatt(wetland, groups, duleg = TRUE,
control = how(nperm=999))
summary(indvalori)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 8
#> Number of species associated to 1 group: 8
#> Number of species associated to 2 groups: 0
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 3
#> stat p.value
#> Ludads 0.907 0.001 ***
#> Orysp. 0.823 0.001 ***
#> Psespi 0.602 0.015 *
#>
#> Group 2 #sps. 1
#> stat p.value
#> Phynod 0.676 0.009 **
#>
#> Group 3 #sps. 4
#> stat p.value
#> Pancam 0.910 0.001 ***
#> Melcor 0.739 0.001 ***
#> Eupvac 0.724 0.001 ***
#> Cynarc 0.602 0.010 **
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1Restricting the order of site groups combinations
The second way to exclude site group combinations from a
multipatt() analysis is to indicate the maximum order of
the combination to be considered. Using the option
max.order we can restrict site group combinations to be,
for example, singletons (max.order = 1, which is equal to
duleg=TRUE), singletons and pairs
(max.order = 2), or singletons, pairs and triplets
(max.order = 3). In the follow example, only singletons and
pairs are considered:
indvalrest <- multipatt(wetland, groups, max.order = 2,
control = how(nperm=999))
summary(indvalrest)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 11
#> Number of species associated to 1 group: 7
#> Number of species associated to 2 groups: 4
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 3
#> stat p.value
#> Ludads 0.907 0.001 ***
#> Orysp. 0.823 0.004 **
#> Psespi 0.602 0.015 *
#>
#> Group 3 #sps. 4
#> stat p.value
#> Pancam 0.910 0.001 ***
#> Eupvac 0.724 0.003 **
#> Cynarc 0.602 0.017 *
#> Abemos 0.447 0.048 *
#>
#> Group 1+2 #sps. 1
#> stat p.value
#> Elesp. 0.741 0.008 **
#>
#> Group 2+3 #sps. 3
#> stat p.value
#> Melcor 0.876 0.001 ***
#> Phynod 0.715 0.011 *
#> Echell 0.651 0.012 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1In this case the output looks like a the output of an unrestricted
multipatt() execution, because the only combination that is
excluded is the set of all sites, which cannot be tested for
significance and thus never appears in the summary.
Limiting the maximum order of combinations is the commonest way to restrict combinations to avoid computational problems derived from a large number of site groups.
Specifying the site groups combinations to be considered
There is a third, more flexible, way of restricting site group
combinations. The input parameter vector restcomb allows
specifying the combinations of site groups that are permitted in
multipatt(). In order to learn how to use parameter
restcomb, we must first understand that inside
multipatt() site groups and site group combinations are
referred to with integers. Site group combinations are numbered starting
with single groups and then increasing the order of combinations. For
example, if there are three site groups, the first three integers ‘1’ to
‘3’ identify those groups. Then, ‘4’ identifies the combination of Group
1 and Group 2, ‘5’ identifies the combination of Group 1 and Group 3,
and ‘6’ identifies the combination of Group 2 and Group 3. Finally, ‘7’
identifies the combination of all three groups.
The numbers composing the vector passed to restcomb
indicate the site groups and site group combinations that we want
multipatt() to considered as valid options. For example, if
we do not want to consider the combination of Group 1 and Group 2, we
will exclude combination ‘4’ from vector restcomb:
indvalrest <- multipatt(wetland, groups, restcomb = c(1,2,3,5,6),
control = how(nperm=999))
summary(indvalrest)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 33
#> Selected number of species: 9
#> Number of species associated to 1 group: 6
#> Number of species associated to 2 groups: 3
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 3
#> stat p.value
#> Ludads 0.907 0.001 ***
#> Orysp. 0.823 0.003 **
#> Psespi 0.602 0.020 *
#>
#> Group 3 #sps. 3
#> stat p.value
#> Pancam 0.910 0.001 ***
#> Eupvac 0.724 0.004 **
#> Cynarc 0.602 0.014 *
#>
#> Group 2+3 #sps. 3
#> stat p.value
#> Melcor 0.876 0.001 ***
#> Phynod 0.715 0.006 **
#> Echell 0.651 0.020 *
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1If we compare these last results with those including all possible site group combinations, we will realize that species ‘Elesp.’ was formerly an indicator of Group 1 and Group 2, and now it does not appear in the list of indicator species. If fact, if we examine the results more closely we see that the highest IndVal for ‘Elesp’ is achieved for group 1, but this relationship is not significant:
indvalrest$sign
#> s.1 s.2 s.3 index stat p.value
#> Abefic 0 1 0 2 0.2672612 0.557
#> Merhed 0 0 1 3 0.4019185 0.197
#> Alyvag 0 1 0 2 0.3347953 0.413
#> Pancam 0 0 1 3 0.9098495 0.001
#> Abemos 0 0 1 3 0.4472136 0.059
#> Melcor 0 1 1 5 0.8757059 0.001
#> Ludoct 0 0 1 3 0.3162278 0.244
#> Eupvac 0 0 1 3 0.7236825 0.004
#> Echpas 0 1 1 5 0.5842649 0.183
#> Passcr 0 0 1 3 0.3162278 0.256
#> Poa2 0 1 0 2 0.2672612 0.579
#> Carhal 1 0 0 1 0.3313667 0.745
#> Dendio 0 0 1 3 0.3162278 0.230
#> Casobt 1 0 0 1 0.2425356 1.000
#> Aesind 0 1 0 2 0.4447093 0.223
#> Cyprot 0 1 1 5 0.5000000 0.067
#> Ipocop 0 1 1 5 0.3535534 0.319
#> Cynarc 0 0 1 3 0.6017217 0.014
#> Walind 1 0 1 4 0.4406873 0.691
#> Sessp. 0 1 1 5 0.5901665 0.693
#> Phynod 0 1 1 5 0.7145356 0.006
#> Echell 0 1 1 5 0.6509834 0.020
#> Helind 0 1 1 5 0.5720540 0.822
#> Ipoaqu 1 0 1 4 0.4053049 0.899
#> Orysp. 1 0 0 1 0.8229074 0.003
#> Elesp. 1 0 0 1 0.5534178 0.659
#> Psespi 1 0 0 1 0.6023402 0.020
#> Ludads 1 0 0 1 0.9074852 0.001
#> Polatt 1 0 0 1 0.4200840 0.160
#> Poa1 0 1 0 2 0.2672612 0.590
#> Helcri 0 1 0 2 0.2672612 0.590
#> Physp. 0 0 1 3 0.3162278 0.233
#> Goopur 0 0 1 3 0.3162278 0.233Restricting site group combinations is also possible with the phi and point biserial coefficients.
Additional functions to estimate and test the association between species and groups of sites
Although multipatt() is a user-friendly function for
indicator species analysis, other functions are also useful to study the
association between species and site groups.
The function strassoc()
Function strassoc() allows calculating a broad hand of
association indices, described in De Cáceres and
Legendre (2009). For example, we can focus on the ‘A’ component
of IndVal:
prefstat <- strassoc(wetland, cluster=groups, func="A.g")
round(head(prefstat),3)
#> 1 2 3
#> Abefic 0.000 1.000 0.000
#> Merhed 0.000 0.192 0.808
#> Alyvag 0.215 0.785 0.000
#> Pancam 0.024 0.148 0.828
#> Abemos 0.000 0.000 1.000
#> Melcor 0.124 0.330 0.546A feature of strassoc() that is lacking in
multipatt() is the possibility to obtain confidence
interval limits by bootstrapping. In this case, the function returns a
list with three elements: stat, lowerCI and
upperCI:
prefstat <- strassoc(wetland, cluster=groups, func="A.g",
nboot.ci = 199)
round(head(prefstat$lowerCI),3)
#> 1 2 3
#> Abefic 0.000 0.000 0.000
#> Merhed 0.000 0.000 0.000
#> Alyvag 0.000 0.000 0.000
#> Pancam 0.000 0.034 0.662
#> Abemos 0.000 0.000 0.000
#> Melcor 0.032 0.255 0.452
round(head(prefstat$upperCI),3)
#> 1 2 3
#> Abefic 0.000 1.000 0.000
#> Merhed 0.000 1.000 1.000
#> Alyvag 1.000 1.000 0.000
#> Pancam 0.087 0.267 0.943
#> Abemos 0.000 0.000 1.000
#> Melcor 0.184 0.400 0.634For example, the 95% confidence interval for the ‘A’ component of the association between ‘Pancam’ and Group 3 is [0.662, 0.943].
The function signassoc()
As we explained before, multipatt() statistically tests
the association between the species and its more strongly associated
site group (or site group combination). By contrast,
signassoc() allows one to test the association between the
species and each group of sites, regardless of whether the association
value was the highest or not. Moreover, the function allows one to test
both one-sided and two-sided hypotheses. For example, the following line
tests whether the frequency of the species in each site group is higher
or lower than random:
prefsign <- signassoc(wetland, cluster=groups, alternative = "two.sided",
control = how(nperm=199))
head(prefsign)
#> 1 2 3 best psidak
#> Abefic 1.00 0.73 1.00 2 0.980317
#> Merhed 0.34 0.64 0.18 3 0.448632
#> Alyvag 1.00 0.29 0.80 2 0.642089
#> Pancam 0.01 0.14 0.01 1 0.029701
#> Abemos 0.70 0.80 0.07 3 0.195643
#> Melcor 0.01 0.92 0.01 1 0.029701The last columns of the results indicate the group for which the p-value was the lowest, and the p-value corrected for multiple testing using the Sidak method.
Determining how well target site groups are covered by indicators
Besides knowing what species can be useful indicators of site groups (or site group combinations), it is sometimes useful to know the proportion of sites of a given site group where one or another indicator is found. We call this quantity coverage of the site group. Determining the coverage of site groups can be useful for habitat or vegetation types encompassing a broad geographic area (De Cáceres et al. 2012), because there may exist some areas where none of the valid indicators can be found.
The function coverage()
The coverage can be calculated for all the site groups of a
multipatt() object using the function
coverage():
coverage(wetland, indvalori)
#> 1 2 3
#> 1 1 1Note that to obtain the coverage we need to input both the community
data set and the object of class multipatt(). In this case
the coverage was complete (i.e. 100%) for site groups ‘1’ and ‘3’. In
contrast, group ‘2’ has a lower coverage because only one species,
‘Phynod’, can be considered indicator of the site group, and this
species does not always occur in sites of the group.
The coverage of site groups depends on how many and which indicators
are considered as valid. Statistical significance (i.e.,
alpha) determined in multipatt() can be used
to determine what indicators are valid, but we can add more requirements
to the validity of indicator species by specifying additional parameters
to the function coverage(). For example, if we want to know
the coverage of our site groups with indicators that are significant and
whose ‘A’ value is equal or higher than 0.8, we can use:
coverage(wetland, indvalori, At = 0.8, alpha = 0.05)
#> 1 2 3
#> 0.8235294 0.0000000 1.0000000Note that, after adding this extra requirement, group ‘2’ has 0% coverage and the coverage of group ‘1’ has also decreased.
The function plotcoverage()
It is possible to know how the coverage changes with ‘A’ threshold
used to select good indicators. This is obtained by drawing the coverage
values corresponding to different threshold values. This is what the
plotcoverage() function does for us:
plotcoverage(x=wetland, y=indvalori, group="1", lty=1)
plotcoverage(x=wetland, y=indvalori, group="2", lty=2, col="blue", add=TRUE)
plotcoverage(x=wetland, y=indvalori, group="3", lty=3, col="red", add=TRUE)
legend(x = 0.1, y=30,
legend=c("group 1","group 2", "group 3"),
lty=c(1,2,3), col=c("black","blue","red"), bty="n")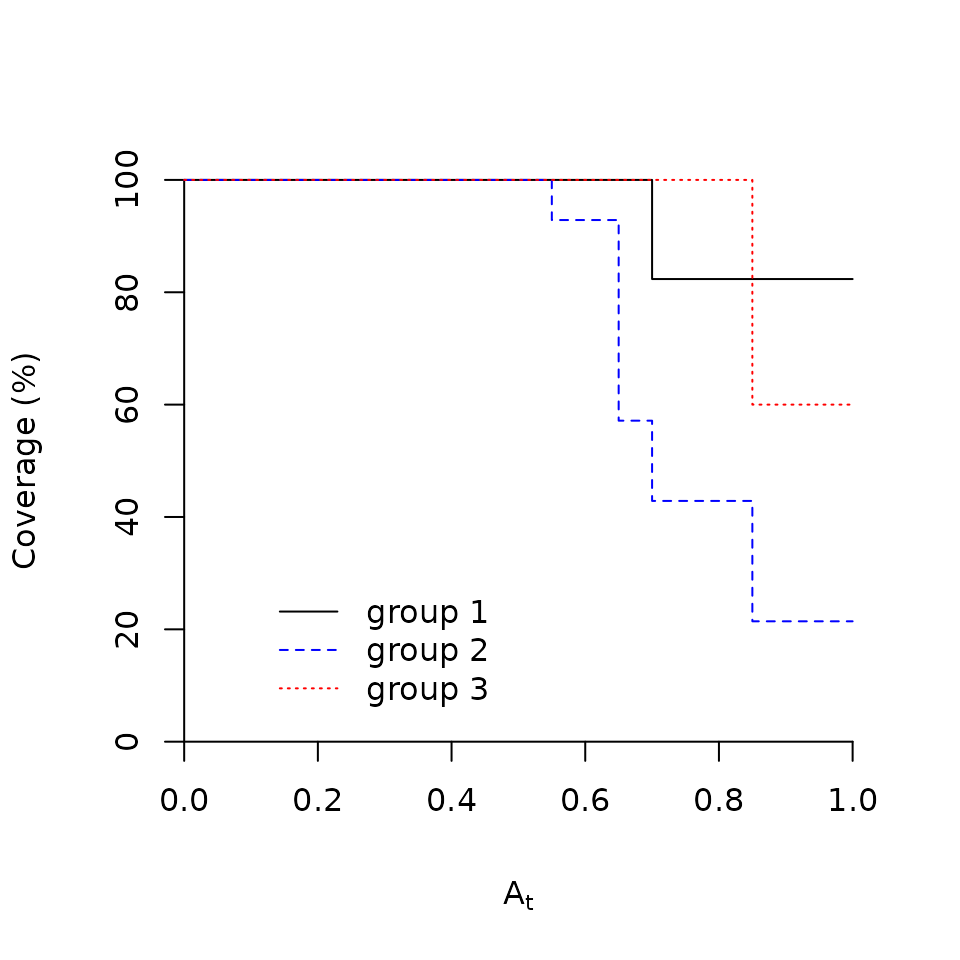
As you can see in the example, function plotcoverage()
has to be called for one group at a time. However, several plots can be
drawn one onto the other using the option add=TRUE.
Species combinations as indicators of site groups
Ecological indicators can be of many kinds. De Cáceres et al. (2012) recently explored the indicator value of combinations of species instead of just considering individual species. The rationale behind this approach is that two or three species, when found together, bear more ecological information than a single one.
Generating species combinations
The association between species combinations and groups of sites is
studied in the same way as for individual species. However, instead of
analyzing a site-by-species matrix, we need a matrix with as many rows
as there are sites and as many columns as there are species
combinations. We can obtain that matrix using the function
combinespecies():
wetcomb <- combinespecies(wetland, max.order = 2)$XC
dim(wetcomb)
#> [1] 41 286The resulting data frame has the same number of sites (i.e. 41) but
as many columns as species combinations (in this case 41 columns). Each
element of the data frame contains an abundance value, which is the
minimum abundance value among all the species forming the combination,
for the corresponding site. In our example, we used
max.order = 2 to limit the order of combinations.
Therefore, only pairs of species were considered. Once we have this new
data set, we can use it in multipatt():
indvalspcomb <- multipatt(wetcomb, groups, duleg = TRUE,
control = how(nperm=999))
summary(indvalspcomb, indvalcomp = TRUE)
#>
#> Multilevel pattern analysis
#> ---------------------------
#>
#> Association function: IndVal.g
#> Significance level (alpha): 0.05
#>
#> Total number of species: 286
#> Selected number of species: 43
#> Number of species associated to 1 group: 43
#> Number of species associated to 2 groups: 0
#>
#> List of species associated to each combination:
#>
#> Group 1 #sps. 14
#> A B stat p.value
#> Orysp.+Ludads 1.0000 0.8235 0.907 0.001 ***
#> Ludads 1.0000 0.8235 0.907 0.001 ***
#> Orysp. 0.6772 1.0000 0.823 0.001 ***
#> Sessp.+Ludads 1.0000 0.4118 0.642 0.001 ***
#> Orysp.+Psespi 1.0000 0.4118 0.642 0.006 **
#> Elesp.+Ludads 1.0000 0.4118 0.642 0.009 **
#> Psespi+Ludads 1.0000 0.4118 0.642 0.006 **
#> Orysp.+Elesp. 0.7424 0.5294 0.627 0.014 *
#> Sessp.+Orysp. 0.9081 0.4118 0.611 0.006 **
#> Psespi 0.8811 0.4118 0.602 0.021 *
#> Helind+Ludads 1.0000 0.3529 0.594 0.004 **
#> Walind+Orysp. 1.0000 0.2941 0.542 0.018 *
#> Walind+Ludads 1.0000 0.2941 0.542 0.018 *
#> Ipoaqu+Ludads 1.0000 0.2941 0.542 0.026 *
#>
#> Group 2 #sps. 11
#> A B stat p.value
#> Phynod+Elesp. 0.9162 0.5714 0.724 0.003 **
#> Phynod 0.6396 0.7143 0.676 0.006 **
#> Phynod+Helind 0.6922 0.6429 0.667 0.006 **
#> Helind+Elesp. 0.6861 0.5714 0.626 0.016 *
#> Phynod+Echell 0.8654 0.3571 0.556 0.017 *
#> Echell+Elesp. 0.8586 0.3571 0.554 0.015 *
#> Melcor+Elesp. 0.7083 0.4286 0.551 0.049 *
#> Aesind+Elesp. 1.0000 0.2857 0.535 0.012 *
#> Echpas+Phynod 0.8293 0.2857 0.487 0.035 *
#> Eupvac+Cyprot 1.0000 0.2143 0.463 0.048 *
#> Echpas+Cyprot 1.0000 0.2143 0.463 0.050 *
#>
#> Group 3 #sps. 18
#> A B stat p.value
#> Pancam 0.8278 1.0000 0.910 0.001 ***
#> Pancam+Melcor 0.7769 1.0000 0.881 0.001 ***
#> Pancam+Echell 1.0000 0.6000 0.775 0.001 ***
#> Eupvac+Echell 1.0000 0.6000 0.775 0.001 ***
#> Pancam+Eupvac 0.7455 0.8000 0.772 0.001 ***
#> Melcor 0.5463 1.0000 0.739 0.001 ***
#> Melcor+Eupvac 0.6648 0.8000 0.729 0.003 **
#> Eupvac 0.6546 0.8000 0.724 0.004 **
#> Pancam+Cynarc 1.0000 0.5000 0.707 0.001 ***
#> Pancam+Sessp. 0.8077 0.6000 0.696 0.001 ***
#> Melcor+Echell 0.7368 0.6000 0.665 0.004 **
#> Melcor+Cynarc 0.8077 0.5000 0.635 0.005 **
#> Cynarc 0.7241 0.5000 0.602 0.010 **
#> Melcor+Sessp. 0.5895 0.6000 0.595 0.022 *
#> Eupvac+Cynarc 0.8485 0.4000 0.583 0.014 *
#> Cynarc+Sessp. 0.7778 0.4000 0.558 0.021 *
#> Cynarc+Phynod 1.0000 0.3000 0.548 0.014 *
#> Cynarc+Echell 1.0000 0.3000 0.548 0.010 **
#> ---
#> Signif. codes: 0 '***' 0.001 '**' 0.01 '*' 0.05 '.' 0.1 ' ' 1The best indicators for both Group 1 and Group 3 are individual
species (‘Ludads’ and ‘Pancam’). However, Group 2 is best indicated if
we find, in the same community, ‘Phynod’ and ‘Elesp’. Note that the
species forming the indicator combination do not need to be good
single-species indicators themselves. In our example ‘Phynod’ is a good
indicator of Group 2 but ‘Elesp.’ is not. As an aside, note that we used
the option duleg=TRUE in this last example, hence excluding
site group combinations, for simplicity.
The function indicators()
In the previous example, there were many combinations of species that were significantly associated any of the site groups. There is another, more efficient, way of exploring the potential indicators for a given target site group. Say, for example, that we want to determine indicators for our Group 2, and we want to consider not only species pairs but also species trios. We can run the indicator analysis using:
sc <- indicators(X=wetland, cluster=groups, group=2,
max.order = 3, verbose=TRUE,
At=0.5, Bt=0.2)
#> Target site group: 2
#> Number of candidate species: 33
#> Number of sites: 41
#> Size of the site group: 14
#> Starting species 1 ... accepted combinations: 0
#> Starting species 2 ... accepted combinations: 0
#> Starting species 3 ... accepted combinations: 0
#> Starting species 4 ... accepted combinations: 3
#> Starting species 5 ... accepted combinations: 3
#> Starting species 6 ... accepted combinations: 24
#> Starting species 7 ... accepted combinations: 24
#> Starting species 8 ... accepted combinations: 27
#> Starting species 9 ... accepted combinations: 34
#> Starting species 10 ... accepted combinations: 34
#> Starting species 11 ... accepted combinations: 34
#> Starting species 12 ... accepted combinations: 34
#> Starting species 13 ... accepted combinations: 34
#> Starting species 14 ... accepted combinations: 34
#> Starting species 15 ... accepted combinations: 45
#> Starting species 16 ... accepted combinations: 49
#> Starting species 17 ... accepted combinations: 49
#> Starting species 18 ... accepted combinations: 49
#> Starting species 19 ... accepted combinations: 49
#> Starting species 20 ... accepted combinations: 53
#> Starting species 21 ... accepted combinations: 66
#> Starting species 22 ... accepted combinations: 69
#> Starting species 23 ... accepted combinations: 72
#> Starting species 24 ... accepted combinations: 73
#> Starting species 25 ... accepted combinations: 73
#> Starting species 26 ... accepted combinations: 73
#> Starting species 27 ... accepted combinations: 73
#> Starting species 28 ... accepted combinations: 73
#> Starting species 29 ... accepted combinations: 73
#> Starting species 30 ... accepted combinations: 73
#> Starting species 31 ... accepted combinations: 73
#> Starting species 32 ... accepted combinations: 73
#> Starting species 33 ... accepted combinations: 73
#> Number of valid combinations: 73
#> Number of remaining species: 14
#> Calculating statistical significance (permutational test)...We can discard species combinations with low indicator values by
setting thresholds for components A and B (in our example using
At=0.5 and Bt=0.2). The parameter
verbose = TRUE allowed us to obtain information about the
analysis process. Note that, by default, the indicators()
function will consider species combinations up to an order of 5
(i.e. max.order = TRUE). This can result in long
computation times if the set of candidate species is not small.
Similarly to multipatt(), we can print the results of
indicators() for the most useful indicators, using:
print(sc, sqrtIVt = 0.6)
#> A B sqrtIV p.value
#> Phynod+Helind+Elesp. 1.0000000 0.5714286 0.7559289 0.005
#> Phynod+Elesp. 0.9000000 0.5714286 0.7171372 0.005
#> Phynod 0.6666667 0.7142857 0.6900656 0.005
#> Phynod+Helind 0.7142857 0.6428571 0.6776309 0.005
#> Helind+Elesp. 0.6428571 0.5714286 0.6060915 0.010
#> Melcor+Phynod+Elesp. 0.8571429 0.4285714 0.6060915 0.005
#> Melcor+Helind+Elesp. 0.8571429 0.4285714 0.6060915 0.005The species combinations are listed in decreasing indicator value
order. Function indicators() also calculates the
statistical significance of indicator combinations, by making an
internal call to signassoc(). In this case, we obtain that
a combination of ‘Phynod’, ‘Helind’ and ‘Elesp.’ is even a better
indicator than ‘Phynod’ and ‘Elesp.’. Note that the ‘A’ and IndVal
values for the pair ‘Phynod’ and ‘Elesp.’ do not exactly match those
obtained before. This is because indicators() uses ‘IndVal’
and not ‘IndVal.g’ as default association statistic. In contrast to
multipatt(), in indicators() association
statistics are restricted to ‘IndVal’ and ‘IndVal.g’.
Although we did not show it here, indicators() can be
run with the option requesting for bootstrap confidence intervals (using
option nboot.ci). This allow us to know the reliability of
the indicator value estimates, which is specially important for site
groups of small size (De Cáceres et al.
2012).
Determining the coverage for objects of class
indicators()
We can determine the proportion of sites of the target site group
where one or another indicator is found, using, as shown before for
objects of class multipatt(), the function
coverage():
coverage(sc)
#> [1] 1In this case the coverage was complete (i.e. 100%). Like we did for
objects of class multipatt(), we can add requirements to
the validity of indicators. For example:
coverage(sc, At=0.8, alpha =0.05)
#> [1] 0.9285714While we do not show here, in the case of objects of class
indicators() we recommend to explore the coverage using the
lower boundary of confidence intervals, using option type
of function coverage().
Finally, we can also plot coverage values corresponding to different thresholds:
plotcoverage(sc)
plotcoverage(sc, max.order=1, add=TRUE, lty=2, col="red")
legend(x=0.1, y=20, legend=c("Species combinations","Species singletons"),
lty=c(1,2), col=c("black","red"), bty="n")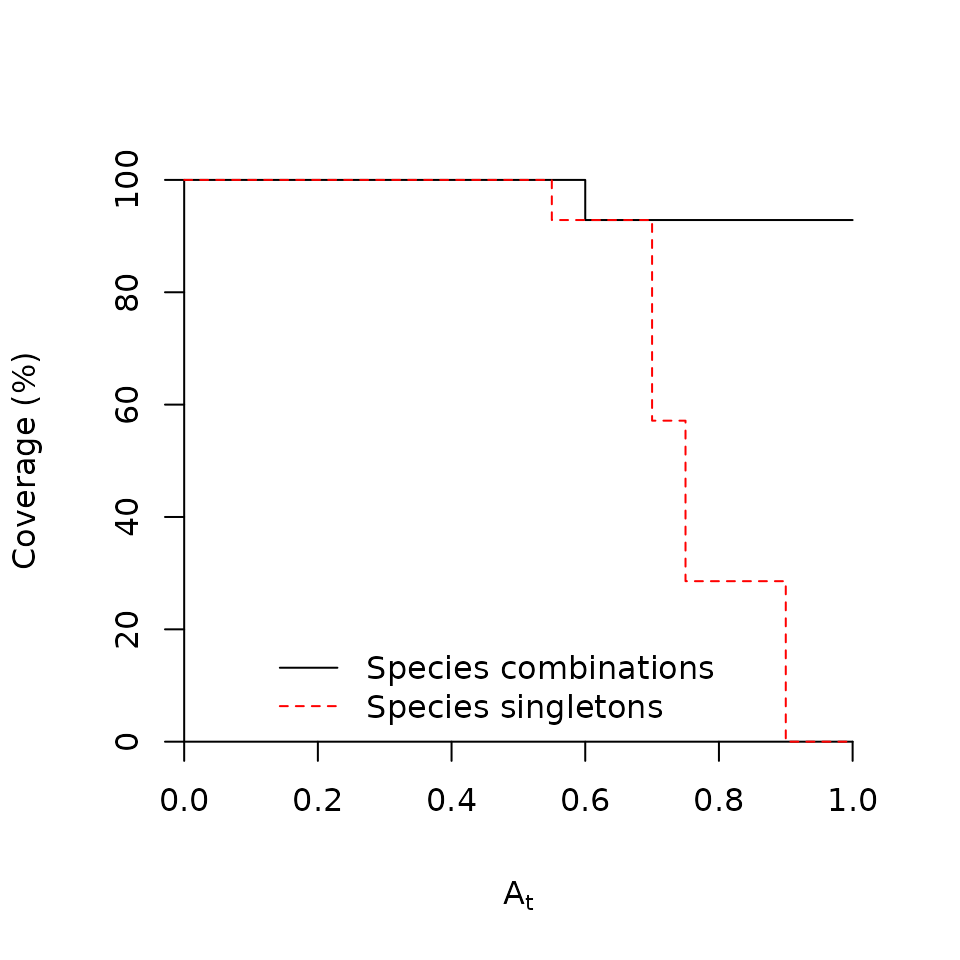
The coverage plot tells us that if we want to use a very large `A’ threshold (i.e., if we want to be very strict to select valid indicators), we won’t have enough valid indicators to cover all the area of our target site group. This limitation is more severe if only single species are considered.
The function pruneindicators()
As there may be many species combinations that could be used as
indicators, the function pruneindicators() helps us to
reduce the possibilities. First, the function selects those indicators
(species or species combinations) that are valid according to the input
thresholds At, Bt and alpha.
Second, the function discards those valid indicators whose occurrence
patterns are nested within other valid indicators. Third, the function
evaluates the coverage of the remaining set of indicators. Finally, it
explores subsets of increasing number of indicators, until the same
coverage as the coverage of the complete set is attained and the subset
of indicators is returned.
sc2 <- pruneindicators(sc, At=0.8, Bt=0.2, verbose=TRUE)
#> Coverage of initial set of 73 indicators: 100%
#> Coverage of valid set of 31 indicators: 92.9%
#> Coverage of valid set of 7 nonnested indicators: 92.9%
#> Checking 7 subsets of 1 indicator(s) maximum coverage: 57.1%
#> Checking 21 subsets of 2 indicator(s).......... maximum coverage: 85.7%
#> Checking 35 subsets of 3 indicator(s)........ maximum coverage: 92.9%
#> Coverage of final set of 3 indicators: 92.9%
print(sc2)
#> A B sqrtIV p.value
#> Phynod+Elesp. 0.9000000 0.5714286 0.7171372 0.005
#> Cyprot 0.8666667 0.2857143 0.4976134 0.025
#> Echpas+Phynod 0.8000000 0.2857143 0.4780914 0.035In our example, and using these thresholds, the best indicators for group ‘2’ would be: (a) the combination of ‘Phynod’ and ‘Elesp.’; (b) ‘Cyprot’ and (c) ‘Echpas’ and ‘Phynod’. The three indicators together cover 93% of the sites belonging to the target site group.
The function predict.indicators()
The function indicators() provides a model that can be
used to predict a target site group. Once a given combination of species
has been found, the corresponding ‘A’ value is an estimate of the
probability of being in the target site group given the combination of
species has been found. Therefore, the set of indicators could be used
to predict the likelihood of the target site group in a new data set.
The function predict.indicators() has exactly this role. It
takes the results of indicators() and a new community data
matrix as input. Given this input, the function calculates the
probability of the target site group for each site. The following code
exemplifies the use of predict.indicators() with the same
data that was used for the calibration of the indicator model (a new
community data matrix should be used in normal applications):
p <- predict(sc2, wetland)Of course, this will return biased estimates, as the data set used to build the model is the same as the target data set for which probabilities are desired. The same would be obtained using:
p <- predict(sc2)which uses the original data set storied in the
indicators() object. If we want to evaluate these
probability estimates in a cross-validated fashion (i.e., excluding each
target site for the calculation of positive predictive values and the
probability of the target site group), we can use:
pcv <- predict(sc2, cv=TRUE)We can compare the probabilities (evaluated by resubstitution and cross-validation) with the original group memberships:
data.frame(Group2 = as.numeric(wetkm$cluster==2), Prob = p, Prob_CV = pcv)
#> Group2 Prob Prob_CV
#> 5 1 0.0000000 0.0000000
#> 8 1 0.0000000 0.0000000
#> 13 1 0.0000000 0.0000000
#> 4 1 0.0000000 0.0000000
#> 17 1 0.0000000 0.0000000
#> 3 1 0.0000000 0.0000000
#> 9 1 0.0000000 0.0000000
#> 21 1 0.0000000 0.0000000
#> 16 1 0.0000000 0.0000000
#> 14 1 0.0000000 0.0000000
#> 2 1 0.0000000 0.0000000
#> 15 1 0.0000000 0.0000000
#> 1 1 0.0000000 0.0000000
#> 7 1 0.0000000 0.0000000
#> 10 1 0.0000000 0.0000000
#> 40 0 0.9000000 1.0000000
#> 23 0 0.0000000 0.0000000
#> 25 0 0.9000000 0.8888889
#> 22 0 0.8000000 0.7500000
#> 20 0 0.9000000 0.8888889
#> 6 0 0.9000000 0.8888889
#> 18 0 0.9000000 0.8888889
#> 12 0 0.9000000 0.8888889
#> 39 0 0.9000000 0.8888889
#> 19 0 0.9000000 0.8750000
#> 11 0 0.9000000 0.8888889
#> 30 0 0.8666667 0.8181818
#> 34 0 0.8666667 0.8333333
#> 28 0 0.0000000 0.0000000
#> 31 0 0.8666667 0.8461538
#> 26 0 0.8666667 0.8181818
#> 29 0 0.8666667 0.9285714
#> 33 0 0.8666667 0.9285714
#> 24 0 0.0000000 0.0000000
#> 36 0 0.0000000 0.0000000
#> 37 0 0.0000000 0.0000000
#> 41 0 0.0000000 0.0000000
#> 27 0 0.0000000 0.0000000
#> 32 0 0.0000000 0.0000000
#> 35 1 0.0000000 0.0000000
#> 38 1 0.0000000 0.0000000Cross-validated probabilities will tend to be lower than probabilities evaluated by resubstitution for sites originally belonging to the target site group and higher for other sites.